TEV Protease Design Contest
Colby Agostino cagostino@wistar.org
Russell Ault aultr@chop.edu
Caroline Davis cada9056@colorado.edu
Sam Garfinkle* Samuel.Garfinkle@pennmedicine.upenn.edu
Michaela Helble Michaela.Helble@pennmedicine.upenn.edu
Andrew Nelson Joseph.Nelson@pennmedicine.upenn.edu
Shahlo Solieva ssolieva@wistar.org
*Corresponding Author
Executive Summary
Our approach to this design competition sought to incorporate data from a variety of sources. To begin, we performed a literature review of available TEV protease mutants and associated design principles, selecting favorable mutations for binding, catalytic activity, and solubility. We then prepared a model of the full-length TEV protease bound to target peptide (ENLYFQG) using previously determined crystal structures, AlphaFold2, and homology modeling. We subjected this model to active site sequence re-design using ProteinMPNN and unbiased molecular dynamics (MD) simulations in both the apo and peptide-bound states to identify putative gatekeeper residues. Integrating this information together, we produced designs that combined previously reported catalytic mutations with previously reported solubility mutations (Design 1), several sets of catalytic mutations and solubility mutations, as well as our sequence re-design results (Design 2), and mutation of MD-identified gatekeeper residues alongside previously described results (Design 3), as well as a final design that truncated the C-terminus to sacrifice specificity for faster diffusion of peptide into and out of the active site (Design 4). Details for each of these steps and designs are outlined below.
Literature Review
We read a number of papers during our literature review, which can be found in the references. We will limit ourselves to discussion of the papers which reported mutations used in our final designs. Note: we report all of our final mutations using a numbering scheme that includes the 6xHIS tag included at the N-terminus, meaning that literature mutations are shifted +7 from their published positions (ex. our S10I is reported as S3I in the literature).
One particularly useful paper was Wang et al.1, which reported a molecular dynamics characterization of two TEV mutants, eTEV (S3I, P8Q, S31T, T173A, V219R, A231V) reported by Denard et al.2 and uTEV3 (I138T, S153N, T180A) reported by Sanchez and Ting3, with kinetic parameters for cleavage of similar peptide substrate ENLYFQSG. eTEV features improvement to Kcat with minimal change to Km, resulting in 3-fold higher efficiency as categorized by Kcat/Km (6.31 vs. 2.23 mM-1s-1 for wild-type with S219V). By contrast, uTEV3 features a similar improvement in Kcat/Km (6.82 vs. 2.23 mM-1s-1 for wild-type with S219V) but achieves this by decreasing Km while leaving Kcat unchanged. We reasoned that, for the purposes of this competition, improvements to Kcat would be more important due to likely excess of target peptide, and we therefore chose eTEV as our top literature design to serve as the platform for further designs. We also decided to use the S219V mutation reported by Kapust et al.4, which was shown to decrease autoproteolysis and thereby increase catalytic activity.
We also found a number of mutations to enhance solubility without sacrificing activity reported in the literature. A paper by Mohammadian et al.5 summarized prior solubilization mutations. They reported a prior double mutant from Cabrita et al.6 (L56V/S135G) with significantly increased solubility compared to wild-type. Additionally, they report a prior triple mutant from van den Berg et al.7 (T17S/N68D/I77V) with 5-fold increased production in E. coli. Finally, they report that Fang et al.8 combined these 5 mutations (L56V/S135G/T17S/N68D/I77V), finding a 5-fold increase in maximum achievable concentration over wild-type. Finally, Hu et al.9 reported that the E106G mutation on a background of the prior 5 solubility mutations was found to rescue the solubility of variants that had been codon optimized for expression in inclusion bodies, suggesting a role for this mutation in solubilization as well. We decided to incorporate all of these solubilizing mutations into our base design given the data supporting their efficacy.
Model Preparation
In order to generate a model of TEV to be used in simulations and interrogation of designs, we ran the full sequence provided for 6xHIS_TEV on AlphaFold210 (AF2) using templates up to 2020-05-14. We then compared the resultant AF2 prediction to chain A of a published TEV crystal structure (PDB 1Q31; TEV C151A bound to peptide MNELVYSQ)11. The top AF2 model (pLDDT ~88) nonetheless had a calculated CA-RMSD to the crystal structure of ~2.5 angstroms, leading us to believe that the crystal structure would serve as a better model for the enzyme core. Therefore, using Free Maestro12, we conjugated the N-terminus and C-terminus of our top AF2 model to the crystal structure, removed the bound peptide, reverted the C151A mutation, pre-processed the protein and optimized hydrogen bonding using the PROPKA tool to generate an apo TEV structure.
As the peptide bound to our crystal structure template was far in sequence space from the peptide chosen for this competition, we used the PDB13 to search for other protease structures with a more similar peptide bound. We found a structure of the homologous protein tobacco vein mottling protease (TVMP) bound to the peptide ETVRFQS (PDB 3MMG)14, which required only 4 mutations to generate the proper peptide sequence of ENLYFQG. We aligned 3MMG to 1Q31 using an alpha carbon (CA) alignment, removed the bound peptide in 1Q31, performed these mutations to the peptide in Free Maestro, and resolved resultant clashes using the built-in minimization tool. We reverted the C151A mutation, pre-processed the protein and optimized hydrogen bonding using the PROPKA tool to generate a target peptide bound TEV structure.
ProteinMPNN redesign
To explore sequence optimization at different distances from the active site, we generated residue selections using PyMOL15 for all enzyme residues containing at least one atom within 3 angstroms, 5 angstroms, or 7 angstroms of the target peptide atoms. We then used ProteinMPNN16 to generate 100 sequences for each of these selections (300 sequences total), and used AF2 to generate predicted structures for each sequence; the results are shown in Figure 1. While many of the predicted structures featured significantly worse pLDDT and CA-RMSD than the WT AF2 prediction, a number were similar to or slight improvements over the WT sequence. We therefore employed cutoffs of pLDDT>87 and CA-RMSD<4.2 angstrom (cutoffs chosen to give ~50 sequences), collecting all 41 sequences meeting these conditions and generating sequence logos to represent the preference of MPNN, shown in Figure 2. While MPNN still preferred the WT sequence at many of these residues, we noticed a number of residues where selected MPNN sequences had an exclusive preference for a residue other than the WT sequence, leading us to believe that these mutations might favor increased stability for the given backbone structure. These mutations were N19T, C26V, H27K, N30T, F44Y, F47Y, L63T, V64I, V70T, F71Y, K72T, V73I, N75D, M89L, M94L, E109V, E113G, I116V, C117V, T121R, T125G, K126G, S129G, S142G, Q152P, S160L, I173L, V189I, E229G, E230P, F232P, A238N, Q240P, N243G. We noticed that many of these mutations were to residues similar in property (hydrophobic/hydrophilc) but slightly different in geometry to the WT sequence, suggesting possible subtle optimization of geometry; others were clearly targeted to accommodate loops or turns using flexible/inflexible residues (substitution of G or P in place of other amino acids, respectively). Inspecting the effect of these mutations individually in Maestro, we selected the following as good candidates for inclusion in later designs, using criteria including packing with nearby residues, new contacts/hydrogen bonds, effect on surface solubility, and lack of conflict with our selected literature mutations: C26V, F44Y, F47Y, V64I, V70T, F71Y, V73I, M89L, M94L, I116V, I173L, V189I.
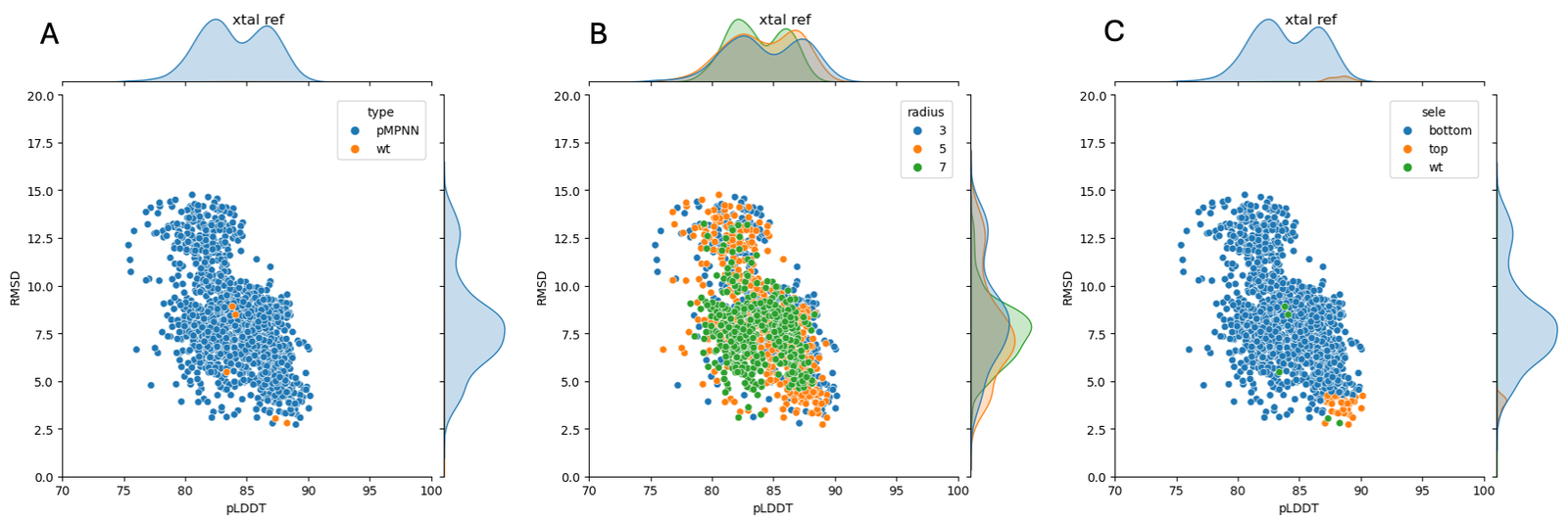


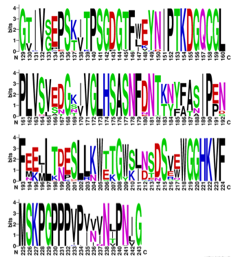

Unbiased MD simulations of apo TEV and peptide-bound TEV
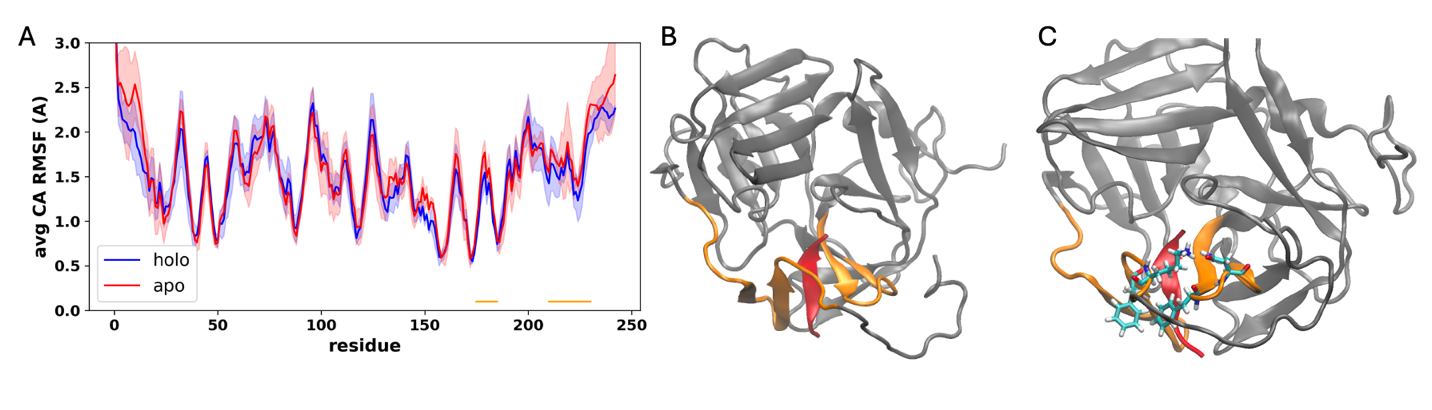
To explore the dynamic effects of certain residues on possible enzymatic activity, we used our apo and peptide-bound TEV models to seed unbiased (“vanilla”) molecular dynamics simulations of the peptide using GROMACS17, running each using 3 replicates for 100ns each, generating a total of 300ns simulation time per design. We then calculated alpha carbon RMSF (CA-RMSF) values for each trajectory, taking an average and standard deviation for each condition and plotting both for comparison (Figure 3A). While the designs were largely similar, we noticed several beta sheets/loops near the active site, underlined/colored in orange, with subtle but notable increases in RMSF in the apo vs. holo simulations, suggesting a stabilizing effect in conjunction with peptide binding. These regions are highlighted in Figure 3B. On visual inspection of the trajectories, it became clear that two phenylalaine residues (F179 and F224) were serving as “gatekeepers” at the active site, while two other residues (N181 and K227) formed a salt bridge at the base of the active site, show in Figure 3C. We hypothesized that mutation of these residues to remove “gatekeeper” effects could speed up catalysis while possibly sacrificing specificity.

Design 1: S10I/P15Q/S38T/T180A/S226R/A238V, T24S/L63V/N75D/I84V/E113G/S142G.
Our first design combined one set of catalytic mutations reported by Denard et al.2 (italicized) with a set of solubilizing mutations (underlined) from several sources mentioned previously4,6–9.
Design 2: Mutations from Design 1 in addition to R226V, I145T, S160N, T187A, C26V, F44Y, F47Y, V64I, V70T, F71Y, V73I, M89L, M94L, I116V, I173L, V189I.
Our second design used all of the previous mutations from design 1, as well as reverting the 226 mutation from R to V (equivalent to S226V mutation to reduce self-cleavage), incorporating the uTEV3 mutations from Sanchez and Ting3 (italicized), and mutations identified using pMPNN that served to optimize packing, folding to requisite backbone shape, and solubility based on our structural inspection (underlined).
Design 3: Mutations from Design 1 in addition to F179A,
F224A, K227S
Our third design used all of the previous mutations from design 1, as well as mutations identified using MD simulations (underlined) that reduce the size of gatekeeper residues and break a critical salt bridge near the active site while preserving solubility, hopefully serving to open it up and allow for faster diffusion of peptide into and out of the active site.
Design 4: Mutations from Design 1 in addition to C-terminus
truncation at P228
Our third design used all of the previous mutations from design 1, and cleaves the C-terminus at residue 228, potentially reducing obstruction of the active site and allowing for faster diffusion of peptide into and out of this site.
Final Designed Sequences:
>Design_1_Wistar_Penn_CHOP_Colorado
MHHHHHHGEILFKGQRDYNPISSSICHLTNESDGHTTTLYGIGFGPFIITNKHLFRRNNGTLVVQSLHGVFKVKDTTTLQQHLVDGRDMIIIRMPKDFPPFPQKLKFREPQRGERICLVTTNFQTKSMSSMVSDTSCTFPSGDGIFWKHWIQTKDGQCGSPLVSTRDGFIVGIHSASNFANTNNYFTSVPKNFMELLTNQEAQQWVSGWRLNADSVLWGGHKVFMRKPEEPFQPVKEVTQLMN
>Design_2_Wistar_Penn_CHOP_Colorado
MHHHHHHGEILFKGQRDYNPISSSIVHLTNESDGHTTTLYGIGYGPYIITNKHLFRRNNGTLVIQSLHGTYKIKDTTTLQQHLVDGRDLIIIRLPKDFPPFPQKLKFREPQRGERVCLVTTNFQTKSMSSMVSDTSCTFPSGDGTFWKHWIQTKDGQCGNPLVSTRDGFIVGLHSASNFANTNNYFASIPKNFMELLTNQEAQQWVSGWRLNADSVLWGGHKVFMVKPEEPFQPVKEVTQLMN
>Design_3_Wistar_Penn_CHOP_Colorado
MHHHHHHGEILFKGQRDYNPISSSICHLTNESDGHTTTLYGIGFGPFIITNKHLFRRNNGTLVVQSLHGVFKVKDTTTLQQHLVDGRDMIIIRMPKDFPPFPQKLKFREPQRGERICLVTTNFQTKSMSSMVSDTSCTFPSGDGIFWKHWIQTKDGQCGSPLVSTRDGFIVGIHSASNAANTNNYFTSVPKNFMELLTNQEAQQWVSGWRLNADSVLWGGHKVAMRSPEEPFQPVKEVTQLMN
>Design_4_Wistar_Penn_CHOP_Colorado
MHHHHHHGEILFKGQRDYNPISSSICHLTNESDGHTTTLYGIGFGPFIITNKHLFRRNNGTLVVQSLHGVFKVKDTTTLQQHLVDGRDMIIIRMPKDFPPFPQKLKFREPQRGERICLVTTNFQTKSMSSMVSDTSCTFPSGDGIFWKHWIQTKDGQCGSPLVSTRDGFIVGIHSASNFANTNNYFTSVPKNFMELLTNQEAQQWVSGWRLNADSVLWGGHKVFMRK
References:
1. Wang,
J., Xu, Y., Wang, X., Li, J. & Hua, Z. Mechanism of Mutation-Induced
Effects on the Catalytic Function of TEV Protease: A Molecular Dynamics Study. Molecules
29, 1071 (2024).
2. Denard, C. A. et
al. YESS 2.0, a Tunable Platform for Enzyme Evolution, Yields Highly Active
TEV Protease Variants. ACS Synth. Biol. 10, 63–71 (2021).
3. Sanchez, M. I. & Ting, A. Y. Directed evolution
improves the catalytic efficiency of TEV protease. Nat. Methods 17,
167–174 (2020).
4. Kapust, R. B. et
al. Tobacco etch virus protease: mechanism of
autolysis and rational design of stable mutants with wild-type catalytic
proficiency. Protein Eng. Des. Sel. 14, 993–1000 (2001).
5. Mohammadian, H., Mahnam, K., Sadeghi, H. M., Ganjalikhany,
M. R. & Akbari, V. Rational design of a new mutant of tobacco etch virus
protease in order to increase the in vitro solubility.
Res. Pharm. Sci. 15, 164 (2020).
6. Cabrita, L. D. et
al. Enhancing the stability and solubility of TEV protease using in silico
design. Protein Sci. Publ. Protein Soc. 16, 2360–2367 (2007).
7. van den Berg, S., Löfdahl,
P.-Å., Härd, T. & Berglund, H. Improved solubility
of TEV protease by directed evolution. J. Biotechnol.
121, 291–298 (2006).
8. Fang, L. et al. An improved strategy
for high-level production of TEV protease in Escherichia coli and its
purification and characterization. Protein Expr. Purif.
51, 102–109 (2007).
9. Hu, J. et al. Combination of the
mutations for improving activity of TEV protease in inclusion bodies. Bioprocess
Biosyst. Eng. 44, 2129–2139 (2021).
10. Evans, R. et al. Protein complex
prediction with AlphaFold-Multimer. 2021.10.04.463034
Preprint at https://doi.org/10.1101/2021.10.04.463034 (2022).
11. Nunn, C. M. et al. Crystal Structure of
Tobacco Etch Virus Protease Shows the Protein C Terminus Bound within the
Active Site. J. Mol. Biol. 350, 145–155 (2005).
12. Schrödinger Release 2023-3: Maestro, Schrödinger,
LLC, New York, NY, 2023.
13. Berman, H. M. et al. The Protein Data
Bank. Nucleic Acids Res. 28, 235–242 (2000).
14. Sun, P., Austin, B. P., Tözsér,
J. & Waugh, D. S. Structural determinants of tobacco vein mottling virus
protease substrate specificity. Protein Sci. 19, 2240–2251
(2010).
15. The PyMOL Molecular
Graphics System, Version 3.0 Schrödinger, LLC.
16. Dauparas, J. et
al. Robust deep learning–based protein sequence design using ProteinMPNN. Science 378, 49–56 (2022).
17. Berendsen, H. J. C., van der Spoel, D. & van Drunen, R.
GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun.
91, 43–56 (1995).