 Approach
1: Combinatorial mutagenesis using evolutionary information and computational
protein design (sequences 1&2)
Approach
1: Combinatorial mutagenesis using evolutionary information and computational
protein design (sequences 1&2)
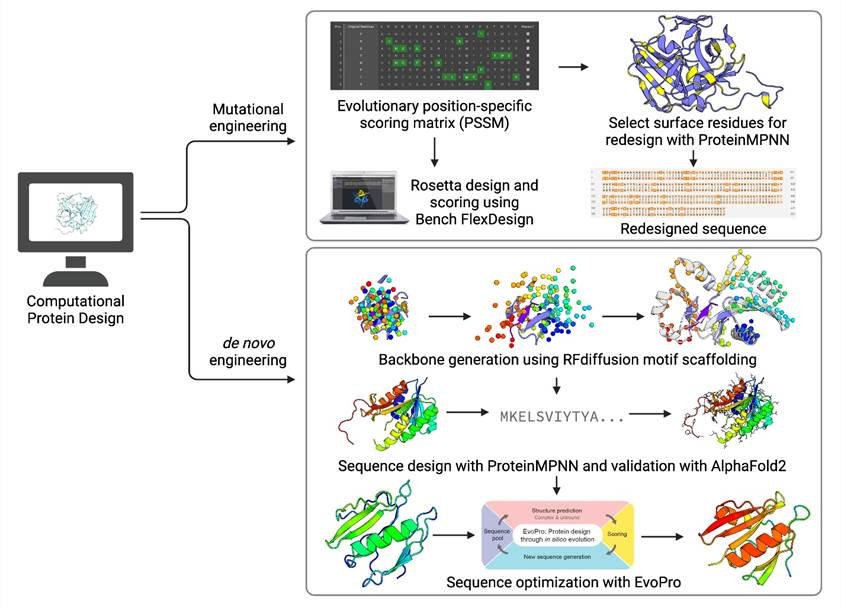
Using the Rosetta GUI by Levitate (Cyrus) Bio, we performed energy minimization on the crystal structure PDB 1LVM followed by site-saturation mutagenesis (SSM). SSM results suggested multiple positions at which amino acid substitutions could improve protein stability (ddG<0), which were selected for combinatorial mutagenesis. These positions are mostly on the surface of TEV protease, which is consistent with the wild-type protein’s low solubility and indicates that the protein surface can be optimized. Levitate also provided a position-specific scoring matrix (PSSM) for TEVp, which highlights the amino acid frequency in homologs of TEV protease at each residue position. We incorporated this evolutionary information into a combinatorial mutagenesis experiment, conducted with the Rosetta FastDesign protocol. Altogether, we sampled both stabilizing mutations and naturally occurring amino acids at 32 positions filtered from SSM on the 1LVM structure. The final design with the lowest free energy score and 31 mutations to the wild-type was selected.
Parallelly, we redesigned the surface of hyperTEV60 (1) using proteinMPNN (2). Surface positions that are evolutionarily conserved were filtered out based on the PSSM, after which they were inputted for proteinMPNN design on the hyperTEV60 backbone. Our final design contains an additional 28 mutations to hyperTEV with increased solubility metrics. The design was also predicted by Rosetta to have lower free energy than hyperTEV.
Approach 2: Active site scaffolding using deep learning
methods (sequences 3&4)
Since the TEV protease backbone has been extensively sampled, we hypothesize that, although less reliable, de novo protein folds could unlock higher improvement in enzyme expression and activity.
Using PDB 1LVM, we determined structural regions that are important for substrate specificity, substrate orientation, and catalysis. These include amino acids in the catalytic triad (His46, Asp81, Cys151) and those making van der Waals or hydrogen-bonding contacts to the peptide substrate. The chosen amino acids were used as motifs in RFDiffusion (3) simulations to scaffold both the active site and the peptide substrate. We experimented with the order of these residues in the simulations and allowed RFDiffusion to construct 10-50 aa backbones in each segment break. Then, we manually assessed and filtered the backbone based on active site RMSD and secondary structures. These backbones were inputted to proteinMPNN for sequence generation and forward folded with AlphaFold2 (4). Sequences were further improved using EvoPro (5), a genetic algorithm coupled with structure prediction pipeline, to ensure good placement of the catalytic triads and high protein folding probability. Although the final sequences are shorter than the wild-type, we hope that it will serve as a proof-of-concept example of minimal but function-preserving scaffolding with RFDiffusion if successful.
Final sequence submission ranked by priority
1.
MHHHHHHGSSLFKGPRDYNPISSTICRLTNRSDGEQTVLYGIGFGPFIITNKHLFRRNNGTLIVQSQHGVFVVPNTTTLEQHLIPGRDMIIIRMPPDFPPFPEKLKFRPPIKDERICLVTTNFQTKSLSSVVSDTSSTVPSSDGIFWQHWIETKDGQCGSPLVSTEDGAIVGIHSASNFTNTNNYFTSVPPNFMELLTNPSAQRWVSGWSLNADSVLWGGHKVFMDKP
2.
MHHHHHHGSSLVPGPRDYNPISDTIVKLTNTSDGETITLYGIGFGPLIITNAHLFRRNNGTLTIESIHGTFVIPNTTTLKLHLIPGRDLVLIEMPEDFPPFPTNLVFRPPVPGEEIVLVTRNFQPKTITSNVSDVSVTRPSSDGVFWEHWIPTKDGQCGSPMVSVKDGSIVGIHSASNFTNTNNYFTAVPENFMELLTDPSLQDWVSGWQLNSESVEWGGHKVFMDKP
3.
MHHHHHHGEVREENGVKRETYLLESEEEARELLDKIIKTKPTKDGQARIIVIAKLPDGRYEVKVITLENLSEEERLEVLEELEEELKKYEEVTIYYHSASNFTNTNNYFTLPRDMSGFEHLRKILAGETAVIKGDVEVYDEKTGKWVLHKNATLVIV
4.
MHHHHHHMGIELYISLNTKDGQAYIYYKGPDGKWYKISFSDLEKKTEVSEEEAEHSASNFTNTNNYFTKKQNRDMLSEEEFEEHLEKLKKGETVEITLKSTGKKVFMSKPMNGKMELVYSQ
References
1. Sumida, K. H., Núñez-Franco, R., Kalvet,
I., Pellock, S. J., Wicky, B. I. M., Milles, L. F., Dauparas, J., Wang, J.,
Kipnis, Y., Jameson, N., Kang, A., De La Cruz, J., Sankaran, B., Bera, A. K.,
Jiménez-Osés, G., and Baker, D. (2024) Improving Protein Expression, Stability,
and Function with ProteinMPNN. J. Am. Chem. Soc. 146, 2054–2061
2. Dauparas, J., Anishchenko, I., Bennett, N.,
Bai, H., Ragotte, R. J., Milles, L. F., Wicky, B. I. M., Courbet, A., de Haas,
R. J., Bethel, N., Leung, P. J. Y., Huddy, T. F., Pellock, S., Tischer, D.,
Chan, F., Koepnick, B., Nguyen, H., Kang, A., Sankaran, B., Bera, A. K., King,
N. P., and Baker, D. (2022) Robust deep learning-based protein sequence design
using ProteinMPNN. Science. 378, 49–56
3. Watson, J. L., Juergens, D., Bennett, N.
R., Trippe, B. L., Yim, J., Eisenach, H. E., Ahern, W., Borst, A. J., Ragotte,
R. J., Milles, L. F., Wicky, B. I. M., Hanikel, N., Pellock, S. J., Courbet,
A., Sheffler, W., Wang, J., Venkatesh, P., Sappington, I., Torres, S. V.,
Lauko, A., De Bortoli, V., Mathieu, E., Ovchinnikov, S., Barzilay, R.,
Jaakkola, T. S., DiMaio, F., Baek, M., and Baker, D. (2023) De novo design of
protein structure and function with RFdiffusion. Nature. 620,
1089–1100
4. Jumper, J., Evans, R., Pritzel, A., Green,
T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A.,
Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. A. A., Ballard, A. J., Cowie,
A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., Back, T., Petersen,
S., Reiman, D., Clancy, E., Zielinski, M., Steinegger, M., Pacholska, M.,
Berghammer, T., Bodenstein, S., Silver, D., Vinyals, O., Senior, A. W.,
Kavukcuoglu, K., Kohli, P., and Hassabis, D. (2021) Highly accurate protein
structure prediction with AlphaFold. Nature. 596, 583–589
5. Goudy, O. J., Nallathambi, A., Kinjo, T.,
Randolph, N. Z., and Kuhlman, B. (2023) In silico evolution of autoinhibitory
domains for a PD-L1 antagonist using deep
learning models. Proc. Natl. Acad. Sci. U. S. A. 120,
e2307371120