Protein Design Games (Winter 2024)
Team #1
Daniella Pretorius, Felipe Engelberger, Ramith Hettiarachchi, Yehlin Cho, Yo Akiyama dp5117@ic.ac.uk, felipeengelberger@gmail.com, ramithhettiarachchi@fas.harvard.edu, yehlin@mit.edu, yo_aki@mit.edu
Design Strategy Overview
Our team aimed to enhance the cleavage activity of the Tobacco Etch Virus (TEV) protease by incorporating mutations that improve thermal stability and solubility. We employed a dual approach, utilizing both the ThermoMPNN and PROSS workflows to identify potential optimizing mutations (Dieckhaus et al., Goldenzweig et al.). A recent study leveraged ProteinMPNN (Sumida et al., J. Dauparas et al.) to increase TEV expression, stability, and function. We decided to use the designed sequence with the highest activity (kcat/Km), which they named HyperTEV60. All designs include the S219V mutation for reduced self-cleavage and an N-terminal 6xHis tag for purification purposes. Aside from optimizing HyperTEV60, we also include a de-novo TEV protease sequence and structure using RFdiffusion and ProteinMPNN.
Design Methods
ThermoMPNN optimization pipeline
To further optimize the HyperTEV60 sequence, we employed ThermoMPNN. ThermoMPNN extracts features from the pre-trained ProteinMPNN model and uses it to train a stability prediction module. The model is trained on a megascale stability dataset (Tsuboyama et al.) to predict changes in thermodynamic stability (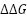 ) of point mutations given the structure and sequence. We intuit that this additional fine-tuning will propose mutations in a semi-orthogonal manner to ProteinMPNN. We optimize the TEV sequence using an iterative workflow with ThermoMPNN (Fig. 1). The workflow takes a starting sequence and structure as input. At each iteration, ThermoMPNN returns predicted changes in thermodynamic stability for all possible point mutations (i.e., site-saturation mutagenesis) (Fig. 2). Our workflow selects the mutation with the single most negative and updates the sequence (Fig. 3).
) of point mutations given the structure and sequence. We intuit that this additional fine-tuning will propose mutations in a semi-orthogonal manner to ProteinMPNN. We optimize the TEV sequence using an iterative workflow with ThermoMPNN (Fig. 1). The workflow takes a starting sequence and structure as input. At each iteration, ThermoMPNN returns predicted changes in thermodynamic stability for all possible point mutations (i.e., site-saturation mutagenesis) (Fig. 2). Our workflow selects the mutation with the single most negative and updates the sequence (Fig. 3).
The design process starts with the HyperTEV60 sequence, and AlphaFold2 (Jumper et al.) predicted structure as input to ThermoMPNN. At each of these iterations, we analyze other metrics described in validation methods and determine which mutations to include. The mutated sequence is then used as the input to ThermoMPNN in the following iteration. Like Sumida et al., we preserve the amino acids of the top 50% of evolutionarily conserved residues and residues within 7Å of the active site.
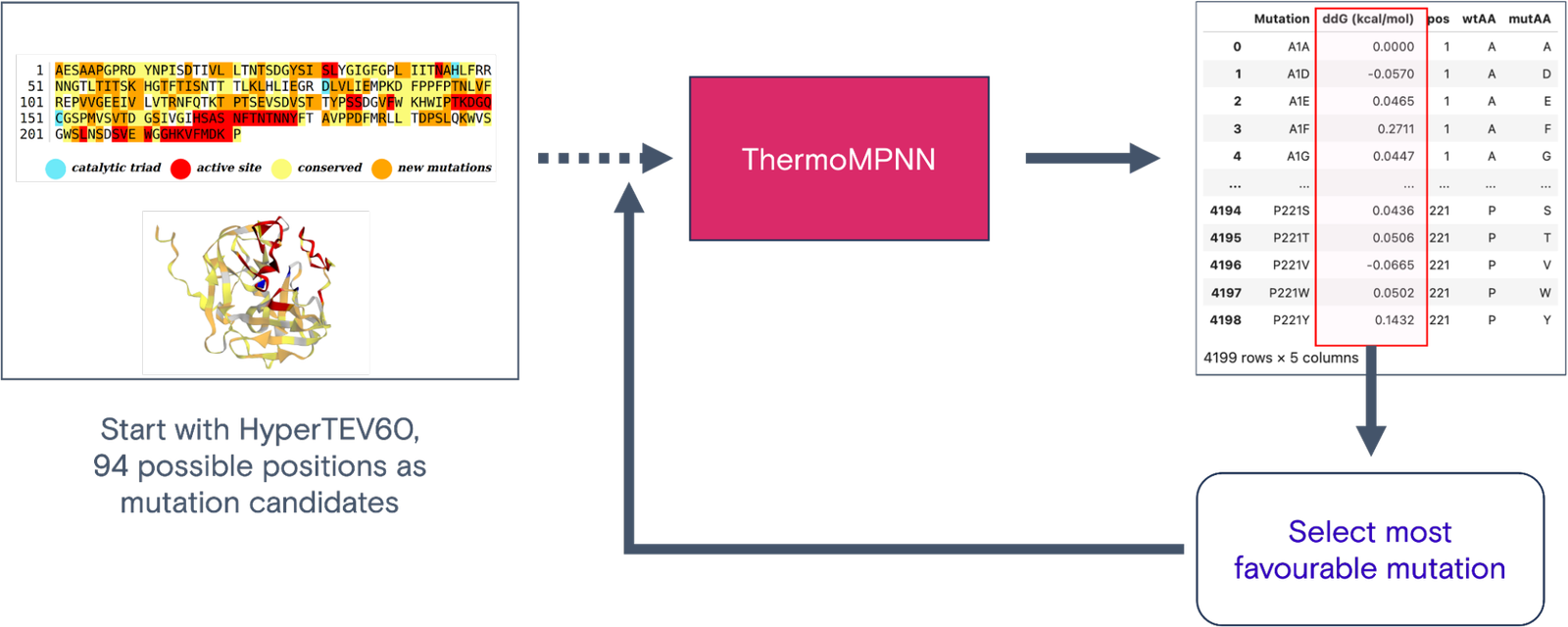
Figure 1: ThermoMPNN-based design pipeline
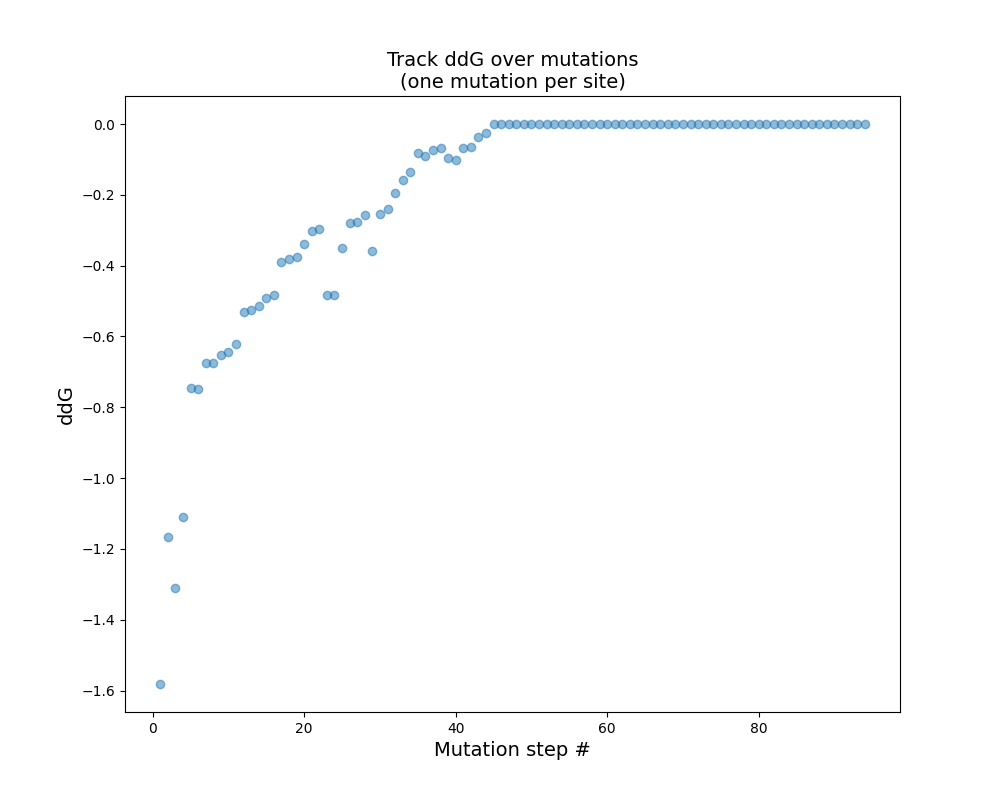
Figure 2: Predicted thermodynamic stability for the top mutation at every iteration
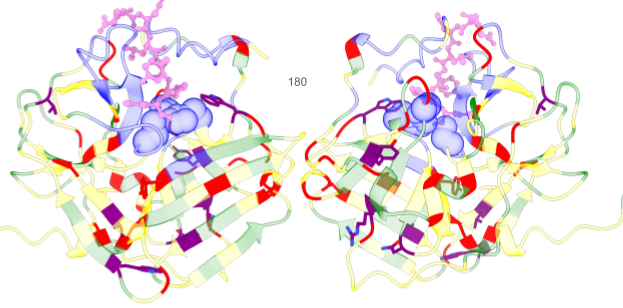
Figure 3. Structure representation of ThermoHyperTEV60. In blue, we highlight the active site residues fixed during the design pipeline; in sphere representation, we show the catalytic triad. In yellow are the 50% conserved residues in Sumida et al. 2023. In green, the mutations to hyperTEV60 were introduced in Sumida et al. 2023, which we kept fixed. In red, we highlight the positions that were not fixed in hyperTEV60 but were not designed by ProteinMPNN. In purple, we highlight the eight mutations our pipeline #X suggested, giving rise to ThermoHyperTEV60. Finally, we superimpose the substrate analog from PDB 1LVM represented with a pink ‘ball and stick’ representation.
PROSS optimization pipeline
We used the PROSS (Protein Repair One Stop Shop) method (Goldenzweig et al., Weinstein et al.), which has previously been shown to be effective at generating variants with a large net stabilizing effect and increased solubility without negatively affecting function. By using the workflow defined below, we use evolutionary information and computational modeling to guide the design of potentially more stable and soluble HyperTEV60 variants (each number corresponds to the workflow segment in Fig. 4):
1. Use Natural Sequence Diversity
- A multiple sequence alignment generates a Position-Specific Substitution Matrix (PSSM). This matrix filters out rare or absent mutations, as determined by evolution.
2. Apply User Determined Constraints
- Fix the top 50% of conserved amino acids in the TEV protease design to maintain critical functional and structural positions.
3. Rosetta Mutational Scanning
- Model each allowed mutation at each allowed position individually to calculate the energy difference (ΔΔG) in Rosetta energy units compared to the original structure. Nine mutation thresholds were tested, from most conservative (ΔΔG ≤ -4) to most permissive (ΔΔG ≤ 0), to assess the impact on protein stability.
4. Rosetta Combinatorial Sequence Design
- Explore combinations of previously identified stabilizing mutations for each mutation threshold to find the optimal combination for enhancing stability.
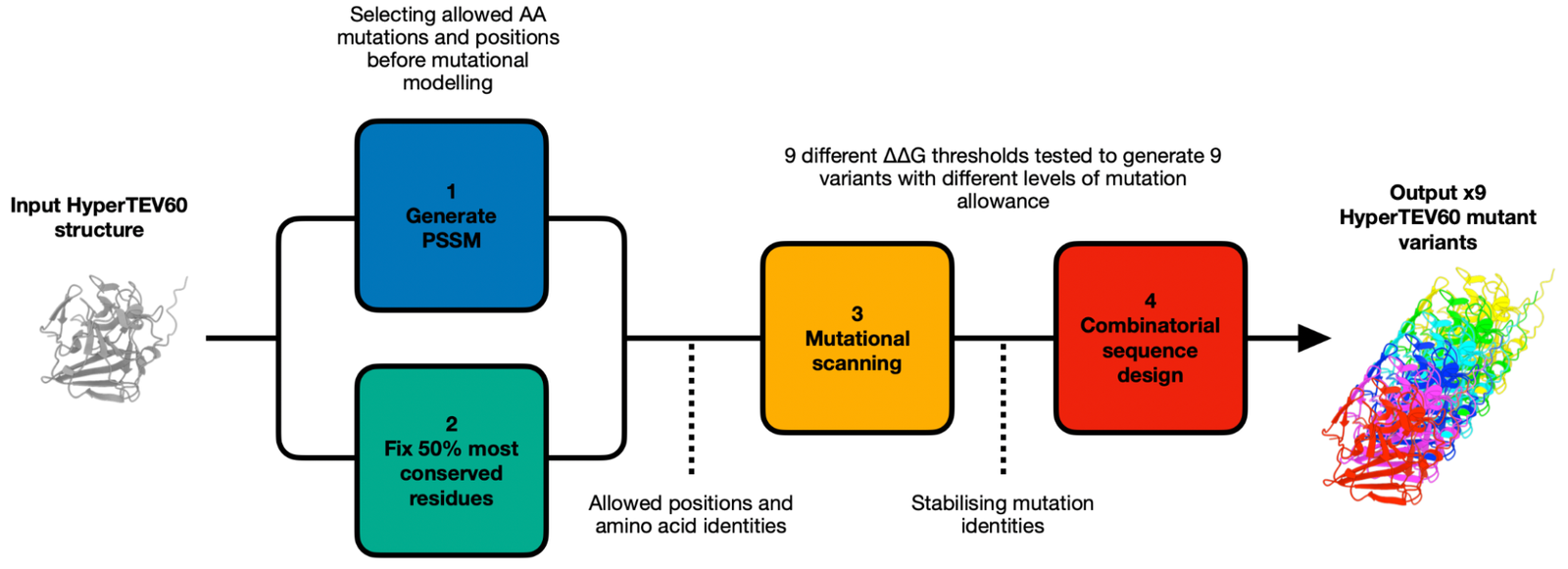
Figure 4: PROSS-based design pipeline
De-novo design using RFdiffusion and ProteinMPNN
For our final design, we used RFdiffusion to construct a de-novo scaffold for the TEV protease catalytic domain (Watson et al.). Here, we fixed the positions and amino acid identities of any residues within 7 Å of the substrate in a ligand-bound complex, and performed partial diffusion (25 noising steps) on all other residues (Figure 5). Partial rather than full diffusion allows us to use HyperTEV60 as a prior for the de-novo design.
We then use ProteinMPNN to design a sequence given the generated structure (Dauparas et al.). We design eight sequences using a temperature of 0.1 and excluding cystines. Importantly, the amino acids near the active site remain unchanged.
Using ESMFold, we measure self-consistency with the RFdiffusion-generated backbone for all 8 ProteinMPNN sequences and select the sequence that results in high pLDDT and low RMSD to the HyperTEV60 catalytic domain.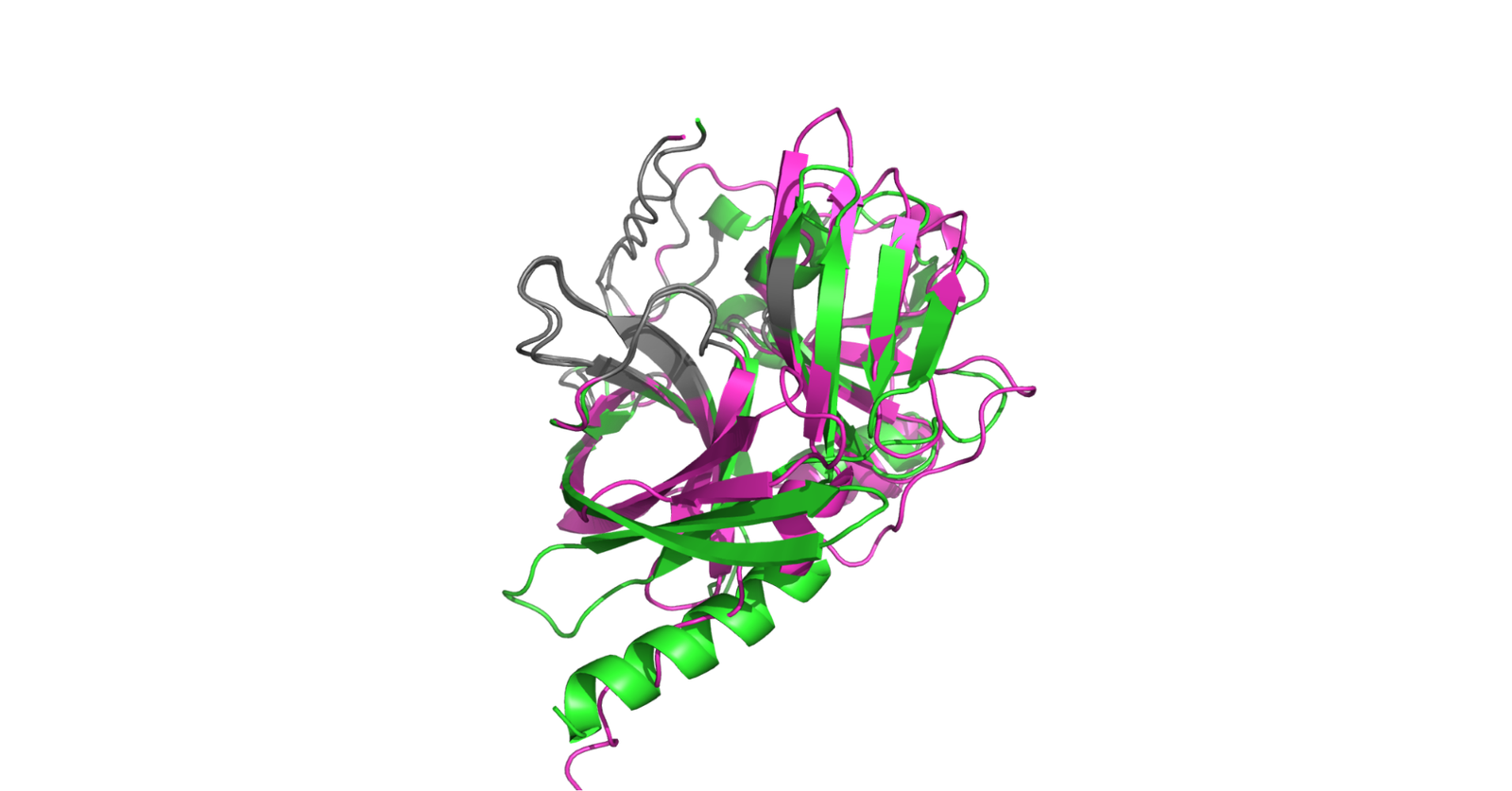
Figure 5. De-novo design superimposed against HyperTEV60. In grey, we highlight the active site residues fixed during the design pipeline. Pink represents the ESMFold predicted structure of HyperTEV60 from Sumida et al. 2023, while green represents the predicted structure of our de-novo design. For the de-novo design, ESMFold predicted pLDDT is 83.2, RMSD to HyperTEV60 is 5.3Å, and catalytic domain RMSD is 2.5Å.
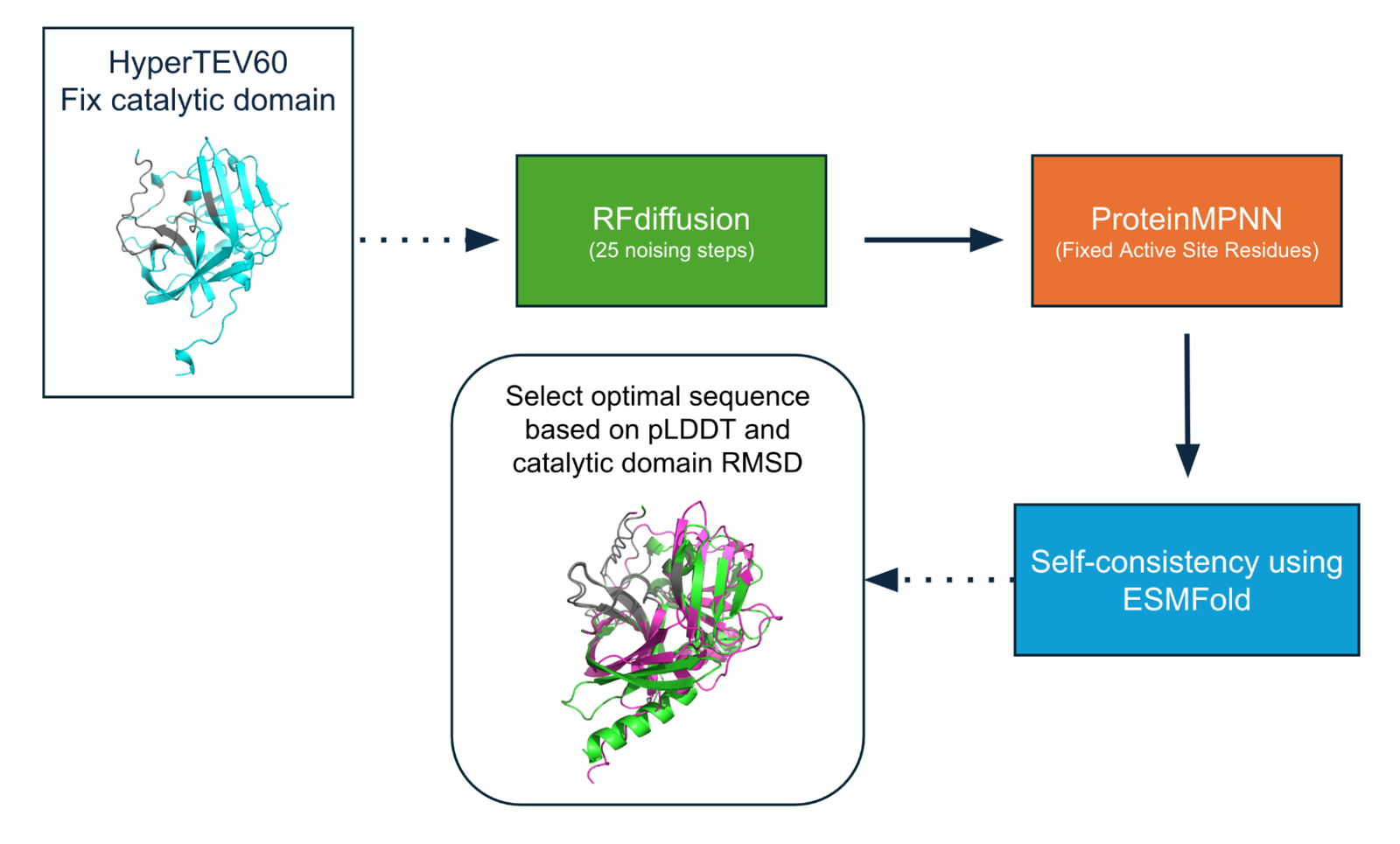
Figure 6: De-novo design pipeline. We fixed the active site starting from HyperTEV60 both in structure (Partial Diffusion) and then we also fixed the active site in the sequence design step (ProteinMPNN).
Validation methods
ESM-2 & ESMfold
We used language model pseudo-perplexity scoring and designability tests to validate our designed sequences to measure their stability and fidelity to the original structure without significant structural changes.
ESM-2 is a protein language model that provides valuable insights into the effects of mutations on protein fold and function. ESM-2 pseudo-perplexity, the exponential of the native pseudo-log-likelihood of a sequence, estimates how well the model predicts the masked tokens of individual tokens in a sequence and enables the prediction of mutation effects. Here, we measured the pseudo-perplexity of designed sequences compared to the wild-type sequence, where lower pseudo-perplexity indicates higher favorability by the ESM-2 model.
In the designability test, we refolded the designed sequences back into their original structures using ESMFold, which is a fully end-to-end single-sequence structure predictor, by training a folding head for ESM-2. We measured pLDDT (predicted Local Distance Difference Test), total RMSD (Root Mean Square Deviation), and active site RMSD for each sequence. Our aim here is to achieve a high model pLDDT and low total RMSD and active site RMSD to maintain similarity to the wild-type structure (Fig. 7).
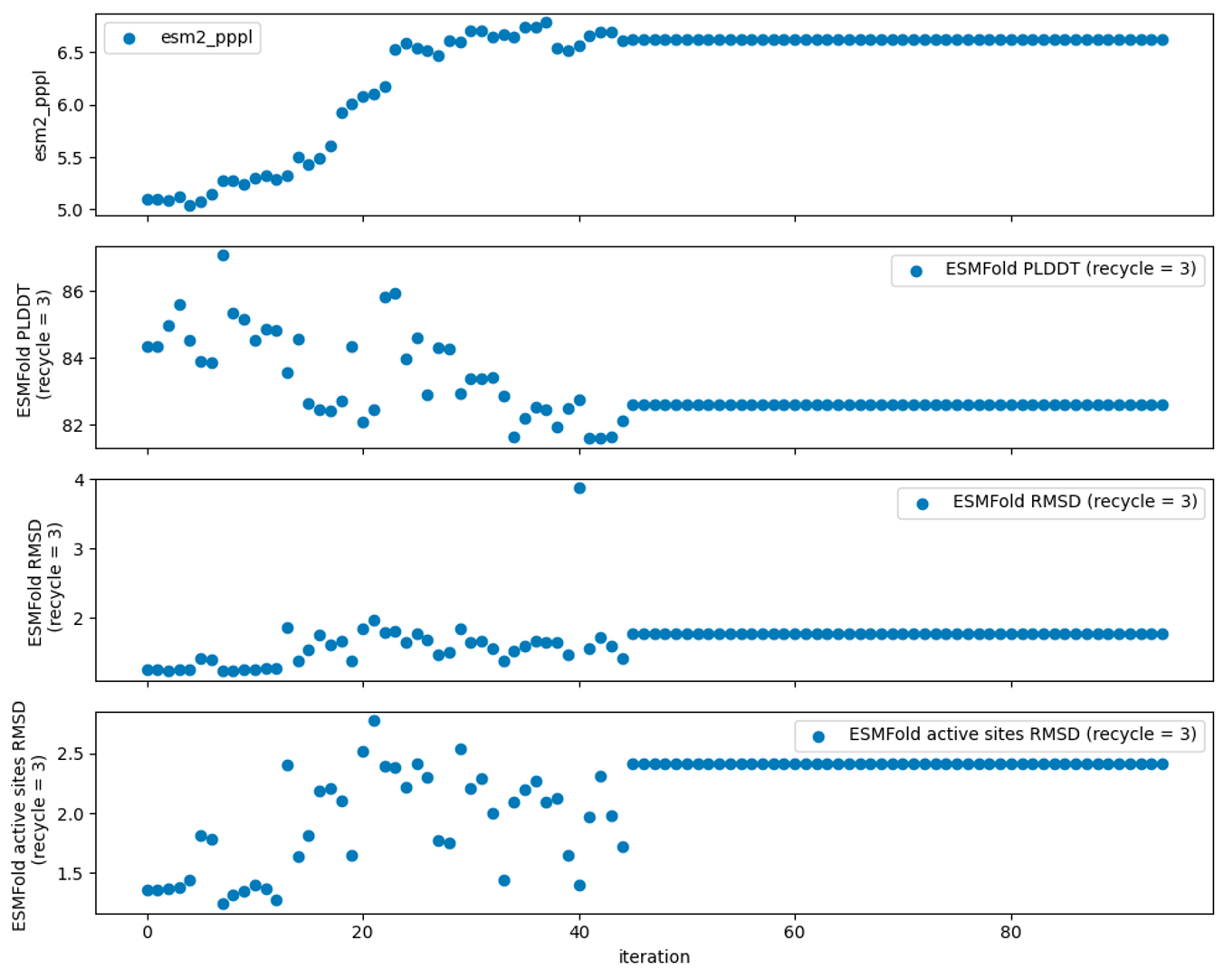
Figure 7: ESM-2 and ESMFold-based validation
Rosetta
We employed the Rosetta(Leman et al. 2020) Energy Breakdown (EB) analysis via the ENDURE pipeline(Engelberger et al. 2023) as an orthogonal validation method for our primary design strategies. This approach allowed us to dissect the energetic contributions of mutations in our designed protein variants. We started by predicting the structures with AF2 for our designed sequences. Then, we ran the PDB files of our designs and the reference TEV protease structure through the ENDURE pipeline so we could systematically prepare our proteins for analysis and execute EB calculations. This process involved structure cleaning, relaxation to a low-energy state using RosettaScripts, and mutation identification. Subsequently, the Rosetta EB analysis provided a detailed view of our designs' energetic landscape, breaking down each residue's pairwise contributions. This granularity enabled us to scrutinize sidechain and backbone interactions, identifying how specific mutations may influence the TEV protease variants' energetic stability.
We used the energy breakdown protocol’s ability to categorize changes in pairwise interactions. It allowed us to understand how mutations affected protein interactions, including indirect effects on non-mutated residues. This comprehensive analysis was pivotal in identifying mutations that either bolstered or detracted from our design goals. Additionally, the built-in residue depth analysis offered insights into the spatial distribution of energetic changes, aiding the differentiation between surface and buried mutations. This analysis was ideal for selecting mutations that improved stability without compromising the protein's stability (Table 1 & 2).
Table 1: ThermoMPNN pairwise mutant summary |
Position | Sum Pairwise Rosetta Energy Breakdown |
39 : P ➡️ N | -0.688 |
49: R ➡️ W | -7.234 |
58: T ➡️ I | 0.786 |
75: H ➡️ Y | -3.265 |
86: E ➡️ R | -1.812 |
114: R ➡️ V | -3.429 |
121 : P ➡️ W | 1.306 |
131: T ➡️ V | -1.826 |
SUM | -16.162 |
Table 2: PROSS pairwise mutant summary |
Position | Sum Pairwise Rosetta Energy Breakdown |
15: S ➡️ A | -0.767 |
19: V ➡️ C | 1.632 |
28: Y ➡️ H | -4.575 |
65: T ➡️ V | -0.973 |
67: S ➡️ K | 0.058 |
71: T ➡️ Q | -0.328 |
78: E ➡️ P | -1.73 |
82: L ➡️ M | -2.671 |
83: V ➡️ I | -0.04 |
86: E ➡️ Q | -3.306 |
96: T ➡️ Q | -1.926 |
97: N ➡️ K | 0.096 |
99: V ➡️ K | 1.211 |
102: E ➡️ Q | 0.036 |
104: V ➡️ K | -1.554 |
105: V ➡️ K | 1.406 |
108: E ➡️ R | -2.27 |
110: V ➡️ C | 2.984 |
128: V ➡️ T | 0.79 |
159: T ➡️ H | 1.388 |
162: S ➡️ C | -0.207 |
185 : D ➡️ N | -0.353 |
188: R ➡️ E | 4.698 |
195: L ➡️ N | 0.199 |
197: K ➡️ E | 0.989 |
SUM | -5.213 |
Design Details
All of the following sequences have mutations suggested by the respective pipeline methods (black) and the previously characterized S219V mutation (purple) (Kapust et al.).
- Seq1: ThermoMPNN Refined Mutations
- Sequence: MHHHHHHAESAAPGPRDYNPISDTIVLLTNTSDGYSISLYGIGFGNLIITNAHLFRRNNGTLTIISKHGTFTISNTTTLKLYLIEGRDLVLIRMPKDFPPFPTNLVFREPVVGEEIVLVTVNFQTKTWTSEVSDVSTVYPSSDGVFWKHWIPTKDGQCGSPMVSVTDGSIVGIHSASNFTNTNNYFTAVPPDFMRLLTDPSLQKWVSGWSLNSDSVEWGGHKVFMVKP
- Strategy: The top 8 mutations with the highest predicted stability increase were selected from the ThermoMPNN workflow previously described. We exclude a mutation R49W after manual review since it is a bulky hydrophobic amino acid on the surface close to the active site.
- Mutations: T131V, R114V, P121W, H75Y, T58I, P39N, E86R, D219V
- Rationale: Thermal stability is often correlated with increased enzymatic activity and solubility, which is crucial for our cell-free expression system.
- Seq2: PROSS Enhanced Specificity Mutations
- Sequence: MHHHHHHAESAAPGPRDYNPISDTIVLLTNTSDGYSISLYGIGFGPLIITNAHLFRRNNGTLTITSKHGTFTIKNTTQLKLHLIEGRDLVLIQMPKDFPPFPTNLVFREPVVGERIVLVTRNFQTKTPTSEVSDVSTTYPSSDGVFWKHWIPTKDGQCGSPMVSVTDGSIVGIHSASNFTNTNNYFTAVPPDFMRLLTDPSLQKWVSGWSLNSDSVEWGGHKVFMVKP
- Strategy: Used the PROSS workflow to identify mutations that enhance stability. Selected the top 4 mutations with a predicted stability increase of at least -1.5 kcal/mol with Rosetta
- Mutations: E86Q, E108R, S67K, T71Q, D219V
- Rationale: These are a selection of the best mutations identified by PROSS over a range of ΔΔG thresholds.
- Seq3: Combined ThermoMPNN and PROSS Mutations
- Sequence: MHHHHHHAESAAPGPRDYNPISDTIVLLTNTSDGYSISLYGIGFGNLIITNAHLFRRNNGTLTIISKHGTFTIKNTTQLKLYLIEGRDLVLIRMPKDFPPFPTNLVFREPVVGERIVLVTVNFQTKTWTSEVSDVSTVYPSSDGVFWKHWIPTKDGQCGSPMVSVTDGSIVGIHSASNFTNTNNYFTAVPPDFMRLLTDPSLQKWVSGWSLNSDSVEWGGHKVFMVKP
- Strategy: Combined mutations identified by both ThermoMPNN and PROSS to leverage the benefits of thermal stability and enhanced specificity. Note that both methods mutate E86, PROSS to the polar Q, and ThermoMPNN to the positively charged R. We select the E86R mutation in this situation, as some of the PROSS variants also returned R in this position.
- Mutations: E108R, S67K, T71Q, T131V, R114V, P121W, H75Y, T58I, P39N, E86R, D219V
- Rationale: This approach aims to create a synergistic effect, potentially leading to a protease variant with superior activity and stability.
- Seq4: top 4 ThermoMPNN + top 4 PROSS
- Sequence: MHHHHHHAESAAPGPRDYNPISDTIVLLTNTSDGYSISLYGIGFGPLIITNAHLFRRNNGTLTITSKHGTFTIKNTTQLKLYLIEGRDLVLIRMPKDFPPFPTNLVFREPVVGERIVLVTVNFQTKTPTSEVSDVSTVYPSSDGVFWKHWIPTKDGQCGSPMVSVTDGSIVGIHSASNFTNTNNYFTAVPPDFMRLLTDPSLQKWVSGWSLNSDSVEWGGHKVFMVKP
- Mutations: T131V, H75Y, R114V, E86R, E108R, S67K, T71Q, D219V
- Strategy and Mutations: We combined the mutations identified by ThermoMPNN and PROSS, limiting the ThermoMPNN mutations to the top 4 for a more conservative design.
- Seq5: Denovo design using RFdiffusion and ProteinMPNN
- Sequence: MHHHHHHSMEEEEIKKKALEILKKAPKMVLLTAPSGHSLWAAVIDNLMIINTHGLSDMKGTFTMTSAGKTKKIDNSFKVVISPLTKNDIVFVKFDEPVEGGGTRYAIVKQPKGKKNVLYKEVETPEEKRIEKLEEFNMSKSSDGIFYEHEMKTKDGQCGTVYYEKGKEDKFYVHSASNFTNTNNYYLAIKESDIEKIKEGKYELAPATIELDVSSVNWGGHKVFMVKP
- Strategy and Mutations: The backbone was designed using partial diffusion on non-catalytic domain residues and the amino acids were assigned using ProteinMPNN. The sequence was selected among seven others based on pLDDT and catalytic domain RMSD.
References
Sumida, Kiera H., et al. "Improving protein expression, stability, and function with ProteinMPNN." Journal of the American Chemical Society 146.3 (2024)
J. Dauparas et al., Robust deep learning-based protein sequence design using ProteinMPNN. Science 378, 49–56 (2022).
H Dieckhaus et al. "Transfer learning to leverage larger datasets for improved prediction of protein stability changes." Proceedings of the National Academy of Sciences 121.6 (2024):
K. Tsuboyama et al., Mega-scale experimental analysis of protein folding stability in biology and design. Nature 620, 434–444 (2023).
A. Goldenzweig et al. Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol. Cell (2016)
Z. Lin et al. "Language models of protein sequences at the scale of evolution enable accurate structure prediction." BioRxiv 2022 (2022)
J.J. Weinstein et al. "PROSS 2: a new server for the design of stable and highly expressed protein variants." Bioinformatics 37.1 (2021)
J. Jumper et al. "Highly accurate protein structure prediction with AlphaFold." Nature 596.7873 (2021)
R.B. Kapust et al. The P1′ specificity of tobacco etch virus protease. Biochem Biophys Res Commun. (2002)
Watson, Joseph L., et al. “De novo design of protein structure and function with RFdiffusion.” Nature 620, 1089–1100 (2023).